Enfermedad de Von Hippel-Lindau y Cáncer renal
ENFERMEDAD DE VON HIPPEL-LINDAU NO FAMILIAR CON CARCINOMA RENAL DE CÉLULAS CLARAS EN EL HOSPITAL REGIONAL DE ALTA ESPECIALIDAD ISSSTE MORELIA. PRESENTACIÓN DE CASO Y REVISIÓN DE LA LITERATURA.
NON-FAMILY VON HIPPEL-LINDAU DISEASE WITH CLEAR CELL RENAL CARCINOMA AT THE ISSSTE MORELIA REGIONAL HIGH SPECIALTY HOSPITAL. PRESENTATION OF THE CASE AND REVIEW OF THE LITERATURE.
Von Hippel-Lindau disease and Renal Cancer.
*Ramsés Rosas Calaña, **Alejandra Galván Ruiz
Revista Oficial del Colegio de Nefrólogos de México AC. ENERO – MARZO, 2021 VOL. 42 No. 1
RESUMEN
La enfermedad de Von Hippel Lindau es ocasionada por mutaciones del gen VHL (un supresor tumoral) que provoca aparición de distintas neoplasias en el sistema nervioso central, ojo, piel y abdomen. Su incidencia es baja, se considera una enfermedad esporádica de carácter hereditario, sin embargo existen casos sin antecedente familiar documentado, lo que dificulta su diagnóstico oportuno; las lesiones oculares son cronológicamente más tempranas y deben hacer pensar en la enfermedad ya que el carcinoma renal de células claras representa la entidad con mayor mortalidad, por lo que su pronta identificación contribuye a mejorar la supervivencia. Presentación del caso: Masculino de 41 años de edad, sin antecedentes heredofamiliares de importancia. Con antecedente de hemangioblastoma en retina y años después en cerebelo. Inició con hematuria total y dolor abdominal. El estudio tomográfico evidenció un tumor renal izquierdo y quistes renales, además de tumoración de cabeza de páncreas y múltiples lesiones quísticas. Se realizó nefrectomía total izquierda, se consignó carcinoma renal de células claras con lo que se confirmó Enfermedad de Von Hippel Lindau no familiar. Continuó su seguimiento para evaluar la función renal y las tumoraciones, así como evaluación de cariotipo para confirmar mutación y valorar consejo genético.
Conclusiones: La enfermedad de Von Hippel Lindau presenta diversas manifestaciones neoplásicas de las cuales el carcinoma renal de células claras representa la entidad con mayor morbimortalidad, por lo que su detección temprana cuando existen antecedentes familiares es esencial para mejorar el pronóstico y diagnosticar a tiempo otras neoplasias vistas en la enfermedad.
Palabras clave: Von Hippel-Lindau, Carcinoma renal, Células claras, Feocromocitoma, Hemangioblastoma
La Enfermedad de Von Hippel-Lindau (EVHL) es una condición hereditaria de componente autosómico dominante de alta penetrancia, que se manifiesta con lesiones de diversa índole, que involucra tejidos derivados del ectodermo (sistema nervioso, células cromafines, ojo y piel) con expresión clínica variable entre los que destacan síndromes neurocutáneos, el más frecuente conocido como facomatosis.
SUMMARY
Von Hippel-Lindau disease is caused by mutations in the VHL gene (a tumor suppressor) that causes the appearance of different neoplasms in the central nervous system, eye, skin and abdomen. Its incidence is low, it has a hereditary nature, however there are cases without a documented family history, which makes its timely diagnosis difficult, the ocular lesions are chronologically earlier and should suggest the disease since, the clear cells represent the entity with the highest mortality, so their early diagnosis improves the survival of patients. Case presentation: 41-year-old male, no important family-inherited antecedents. With a history of hemangioblastoma in the retina and years later in the cerebellum. It begins with total hematuria and abdominal pain. Tomographic study shows left renal tumor and renal cysts, as well as a tumor of the head of the pancreas and multiple cystic lesions. Left total nephrectomy was performed, clear cell renal carcinoma was recorded, confirming non-familial Von Hippel Lindau disease. He continues to be followed up for evaluation of kidney function and tumors, and is sent for karyotype evaluation to confirm mutation and genetic counseling.
Conclusions: Von Hippel Lindau disease presents various neoplastic manifestations of which clear cell renal carcinoma represents the entity with the highest morbidity and mortality, so its early detection when there is a family history is essential to improve prognosis and diagnose in time other neoplasms seen in the disease.
Key words: Von Hippel-Lindau, Renal carcinoma, Clear cells, Pheochromocytoma, Hemangioblastoma
ANTECEDENTES
La Enfermedad de Von Hippel-Lindau (EVHL)es una condición hereditaria de componente autosómico dominante de alta penetrancia, que se manifiesta con lesiones de diversa índole, que involucra tejidos derivados del ectodermo (sistema nervioso, células cromafines, ojo y piel) con expresión clínica variable entre los que destacan síndromes neurocutáneos, el más frecuente conocido como facomatosis. Es causada por la mutación genética el gen VHL en el cromosoma 3p. La EVHL manifiesta la formación de múltiplestumores benignos y malignos, así mismo quistes en múltiples órganos; frecuentemente desarrolla hemangiomas en retina y sistema nervioso central, carcinomas renales de células claras, quistes en epidídimo, feocromocitoma, tumores neuroendócrinos pancreáticos y tumores de saco endolinfatico.3 Su descubrimiento se atribuye a Eugen Von Hippel quien en 1885 describió la angiomatosis retinal congénita; posteriormente, en 1926, Arvin Lindau enlazó los componentes retinal, cerebral y visceral en una sola entidad, quien la describió con alta morbimortalidad. La incidencia de EVHL alcanza un rango de 1 en 36,000 a 45,000 nacimientos y tiene un punto de prevalencia de 1 en 38,000 a 91,000 personas.4 Típicamente la enfermedad se manifiesta en la segunda década de la vida, al momento de la detección, 50% de los pacientes cursan sintomáticos. Los hemangiomas cerebelosos (35%) son comunes. Se ha reportado evidencia de presentación más severa y a temprana edad en generaciones sucesivas.5 La esperanza de vida para pacientes con EVHL históricamente ha estado entre los 40 y 52 años, la expectativa de vida masculina es de 59.4 años, significativamente mayor que la femenina, reportada en 48.1 años. Reportes recientes han establecido la relación de los tumores del sistema nervioso central, la presencia de hemangioblastomas o tumores de células claras del riñón, se relacionan directamente con la expectativa de vida.
El gen VHL (un supresor tumoral), es parte de un complejo multiproteico que incluye elonguina B, elonguina C y culmina 2 (cul2). Este complejo es responsable de la ubiquitinación y degradación de las subunidades alfa de los factores indecibles por hiposa 1 y 2. En hipoxia las propinas no pueden modificarse y pVHL no puede unirse a su ligando, igual que cuando se encuentra ausente o mutando activa la respuesta del gen hipóxico que desencadena la transcripción de un gran repertorio de genes implicados en procesos de angiogénesis, proliferación celular, apoptosis y de metabolismo celular. También se presenta aumento de la expresión de factores tumorales que se asocia con el desarrollo de neoplasias altamente vascularizadas (por crecimiento desmedido del Factor de Crecimiento Endotelial
Vascular, VEFG), observadas en esta entidad.
CASO CLÍNICO
Hombre de 41 años de edad originario del estado de Michoacán, sin antecedentes heredofamiliares o diagnósticos previos de EVHL, con antecedente de resección de tumoración ocular izquierda a los 17 años de edad, y diagnóstico de hemangioblastoma cerebeloso a los 30, manifestado por vértigo y cefalea; el cual requirió extirpación quirúrgica y la colocación de un sistema de derivación ventriculoperitoneal. Inició el padecimiento actual 22 días antes de su ingreso hospitalario, presentó hematuria macroscópica asintomática y se identificó tumoración palpable en flanco izquierdo, por lo que fue valorado por Urología. La exploración física encontró una cicatriz parietal abdominal por cirugía el antecedente quirúrgico, abdomen blando y depresible, doloroso a la palpación de forma intermitente en flanco izquierdo, el cual se irradia hacia la fosa iliaca izquierda, signo de Giordano positivo del lado izquierdo, peristalsis presente sin alteraciones, sin identificar visceromegalias. Extremidades con pulsos periféricos presentes, adecuado llenado capilar, sin limitación al movimiento de extremidades pélvicas. Se realizó tomografía computarizada en fase simple y contrastada (Figura 1) con hallazgos de ambos riñones de localización y situación normal, asimétricos, con múltiples masas hipodensas, bien delimitadas en numero de 12 y 16 en riñones derecho e izquierdo respectivamente de dimensiones entre 1 y 2 cm de diámetro cada uno, compatibles con quistes simples. El riñón izquierdo presentó una masa de densidad y bordes irregulares, bien definidos, que involucra la mitad inferior del riñón izquierdo con diámetros de 95 x 85 x 85 mm, en fase de captación mas hiperdensa y persiste de densidad heterogénea sin evidencia de calcificaciones en su interior, ni de invasión a estructuras adyacentes o involucro de vena renal; la cual desplaza y deforma al sistema colector. La glándula pancreática presentó aumento difuso de sus dimensiones, con presencia de múltiples quistes simples, el mayor de 25 mm de diámetro y a nivel de la cabeza pancreática, una masa sólida, ovoide, de bordes irregulares, bien definidos de 20 mm de diámetro axial que en fase arterial presenta aumento difuso de la densidad, e isodensa en fase de eliminación, compatible con tumor vasculari-zado en estudio; el resto de estructuras abdominales sin evidencia de masas o lesión orgánica en este estudio.
Se realiza estudio complementario sin evidencia de complicación agregada. Con los hallazgos se planeó y se realizó nefrectomía radical izquierda laparoscópica sin complicaciones. La tumoración extraída se envió a estudio histopatológico. No se intervino el proceso quístico pancreático. La evolución fue favorable y el paciente egresó por mejoría. El reporte histopatológico fue de un carcinoma renal papilar de células claras con dimensiones 109 x 80 mm, con zonas de necrosis y hemorragia en 60% del tumor, de un patrón mixto, quístico, tubular y nodular, Furhman II. Sin lesión capsular, borde quirúrgico sin lesión y pielonefritis crónica inespecífica moderada.
Figura 1. Tomografía computarizada corte longitudinal.
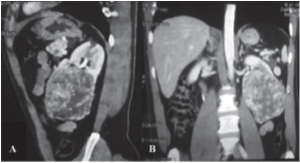
Nota: A, corte longitudinal y B corte coronal, se observa gran tumoración renal izquierda con diferentes densidades y calcificaciones que hacen sospechar proceso maligno.
DISCUSIÓN
Los criterios diagnósticos clínicos para EVHL fueron propuestos por Melmon y Rosen en 1964. Si existe historial familiar de hemangioblastoma en retina o en Sistema Nervioso Central, sólo un hemangioblastoma o lesión visceral (tumor renal, quiste o tumor pancreático, feocromocitoma o cistoadenoma papilar del epidídimo) es necesario para hacer diagnóstico de EVHL. En casos aislados donde no es claro el historial familiar, como en este caso, dos o más hemangioblastomas o un hemangioblastoma y una manifestación visceral es requerido para hacer diagnóstico. Algunos pacientes exhiben todas las manifestaciones de EVHL y en 50% aproximadamente sólo presentan una manifestación.
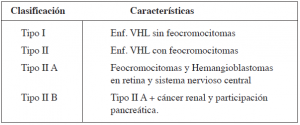
El Instituto Nacional de Cáncer (NCI), ha caracterizado tres fenotipos de la enfermedad de VHL.9 (Tabla 1). Las lesiones de retina generalmente son las primeras en diagnosticarse. La edad media de diagnóstico del hemangioblastoma de retina es a los 25 años de edad; el hemangioblastoma cerebeloso, a los 30 y el carcinoma renal entre los 36 y 44 años. No existe límite de edad para el diagnóstico de EVHL, sin embargo, es inusual el diagnóstico después de los 60 años. La edad media de mortalidad de EVHL es a los 49 años.10 La enfermedad renal es bilateral en al menos 75% de los pacientes, los quistes renales se presentan en 59% a 63% de los pacientes y algunos estudios sustentan que puede aumentar hasta el 85%; estos generalmente anteceden a la detección de tumores sólidos en casi 3 a 7 años. El carcinoma renal se ha reportado en 24 a 45% de los pacientes. La asociación entre carcinoma renal con EVHL exhibe una proporción hombre:mujer casi igual que difiere del carcinoma renal esporádico (mayor en género masculino). 10,11 Las lesiones en EVHL tienden a ser múltiples, bilaterales y con un riesgo elevado de recurrencia.
Los tumores renales sólidos asociados observan una tasa de crecimiento de 0.2 a 2.2 cm/año (media, 1.6 cm/año), la cual es mayor que la encontrada en un carcinoma renal esporádico. Estudios predictivos, mencionan que posiblemente hubiera 600 tumores solidos renales y mas de mil quistes por riñón.11 El tratamiento de los tumores renales requieren intervención quirúrgica, en relación a la clasificación tumoral se asocia terapia neoadyuvante, adyuvante o concomitante. El uso de Pazopanib induce regresión sustancial en hemangioblastomas resistentes a sunitinib.
Pazopanib también se emplea con éxito en el tratamiento de carcinoma renal, principalmente como terapia neoadyuvante. 12,13 El mejor manejo se balancea entre los objetivos de minimizar el riesgo de metástasis de carcinoma renal y preservar la función renal, y los intentos de minimizar el número total de cirugías que un paciente va a requerir a lo largo de su vida. Las opciones disponibles en las formas hereditarias de cáncer renal son la observación con estudios periódicos de imagen cada 6 a 12 meses para monitorización del crecimiento del tumor. La nefrectomía parcial o total, (Nephron Sparing Surgery; NSS) son terapias para evitar metástasis y preservar la función renal en la medida de lo posible. Existen en estudio nuevas terapias como la crioterapia y ablación por radiofrecuencia, sin embargo, aun sin evidencia sólida suficiente. El trasplante renal es una posibilidad en este grupo de pacientes, requiere moni-toreo de la evolución de las lesiones y estar libre de actividad tumoral con un periodo de latencia de entre 2 a 5 años, dependiendo de la estirpe histológica. El uso de inhibidores de Mtor muestra mayor tasa de éxito. Se requiere mas casos y estudio para mejorar las terapias tanto para el injerto como para la EVHL.
Tabla 1. Clasificación de la Enfermedad de Von Hippel Lindau
CONCLUSIONES
La Enfermedad de Von Hippel Lindau es un trastorno genético relacionado con mutaciones en genes supresores de tumores, que como expresiónn clínica desarrolla múltiples neoplasias, de las cuales el carcinoma renal de células claras es el que presenta mayor morbimortalidad. Su diagnóstico es complejo cuando se desconoce el antecedente familiar, hay dificultad de acceso a atención médica especializada o cuando no existen síntomas tempranos que obliguen a buscar atención medica; por lo que se considera una entidad de baja incidencia y alto impacto en morbimortalidad. Deben de crearse estrategias económicas y eficaces enfocadas a este tipo de población para detectar de manera precoz las neoplasias con mayor morbimortalidad, ofertar tratamiento menos invasivos y curativos, al mismo tiempo estudiar a familiares que puedan estar afectados, para de esta manera establecer un protocolo de seguimiento y vigilancia estrecha.
REFERENCIAS
- Zhang L, Wu HK, Wen PY; et al. Increased mobilisation of circulating endothelial progenitors in von Hippel-Lindau disease and renal cell carcinoma. British Journal of Cancer 2011;105(1):112-117.
- Perlman S. Chapter 53. Von Hippel-Lindau disease and Sturge–Weber syndrome. Handbook of Clinical Neuro-logy 2018;148:823-826.
- Cabrera López C. Enfermedades genéticas tumorales renales. Nefrología 2011;(2):1-119
- Rojas Barrantes EI. Enfermedad de Von Hippel Lindau. Revista Médica de Costa Rica y Centroamérica 2013;LXX (605):181-184.
- Salazar R, González CC, Rozas P, Castro J. Retinal capillary hemangioma and von Hippel-Lindau disease: diagnostic and therapeutic implications. Arch Soc Esp Oftalmol 2011;86(7):218-21.
- Clifford SC, Cockman ME, Smallwood AC, Mole DR, Woodward ER, Maxwell PH; et al. Contrasting effects on HIF-1α regulation by disease-causing pVHL mutations correlate with patterns of tumourigenesis in von Hippel-Lindau disease. Hum Mol Genet 2001;1;10(10):1029-38.
- Russell RL, Gladys MG, McClellan W, Emily YC, Steven KL, W Marston L; et al. Lancet 2003;361:2059-67.
- Martínez VD, Jiménez RE, García MR, Barker AA, Dávila RE, García EJ. Tumor renal de células claras como debut de la enfermedad de Von Hippel-Lindau: reporte de caso. Revista Mexicana de Urología 2020;80(1):1-8.
- Schmid S, Gillessen S, Binet I, Brändle M, Engeler D, Greiner J; et al. Management of von Hippel-Lindau Disease: An Interdisciplinary Review. Oncol Res Treat 2014;37(12):761-71.
- Chew EY. Ocular manifestations of Von Hippel- Lindau disease: clinical and genetic investigations. Trans Am Ophthalmol Soc 2005;103:495-511.
- Walther MM, Lubensky IA, Venzon D, Zbar B, Linehan WM: Prevalence of microscopic lesions in grossly normal renal parenchyma from patients with von Hippel-Lindau disease, sporadic renal cell carcinoma and no renal disease: clinical implications. J Urol 1995;154(6):2010-2014.
- Neumann HPH, Young WF and Eng Ch. Pheochromocytoma and Paraganglioma. The New England Journal of Medicine 2019;381(6):552-565.
- Pantigozo RA, Murillo DG, Carreazo NY, Cucho DV. Von Hippel-Lindau disease with extramedullary and pancreatic involvement. Medical Journal Armed Forces India 2020;1-3.
- Jonasch E, McCutcheon IE, Gombos DS, Ahrar K, Perrier ND, Liu D; et al. Pazopanib in patients with von Hippel- indau disease:a single-arm, single-centre, phase 2 trial. The Lancet 2018;1-9.
- Gossage L, Eisen T. Alterations in VHL as potential biomarkers in renal-cell carcinoma. Nat Rev Clin Oncol 2010;7(5):277-288.
- Goldfarb DA, Neumann HP, Penn I, Novick AC. Results of renal transplantation in patients with renal cell carcinoma and von Hippel-Lindau disease. Transplantation 1997;64(12):1726-1729.


Responses